�Ї����w�W(w��ng)Ӎ
������B
�͜؇��ؓp������x��늳ص����ܣ������x��늳���Ҫ���Ќ������Է���,���x�ӔUɢ�ٶȿ��Ó�܄������^�͵�늽��|,�����@������P�I������Li+���܄�����֮�g�����غ͵ăȲ�����ã��@���̘I(y��)̼�၆������늽��|�к��y���F(xi��n),��
���IJ���
�ɹ�����
����,�����ϴ�W���^�ڽ���,�������˵ͽ�늳���(sh��)(��)�܄�������λ���܄����Y������ͨ�^�ʻ������ؓ���{��(ji��)̼�၆��������λ����,������늽��|��−90 ��C�r���F(xi��n)�����x��늌���(1.46 mS��cm−1) ,������−110 ��C�r����Һ�B(t��i)�����,��4.5 Vʯī��ܛ��늳���−10 ��C��200��ѭ�h(hu��n)���_���s98%������ ������,���]���֦�����@Щ늳���−70 ��C�rҲ�ܱ��ּs60%���Ҝط������,����ʹ�ڼs−100 ��C�Ĝض���Ҳ�����E��ر��ַ�늹���,��ԓ�о����}Ŀ�顰Breaking solvation dominance of ethylene carbonate via molecular charge engineering enables lower temperature battery����Փ�İl(f��)���ڇ��H피��ڿ���Nature Communications���ϡ�

�D���x
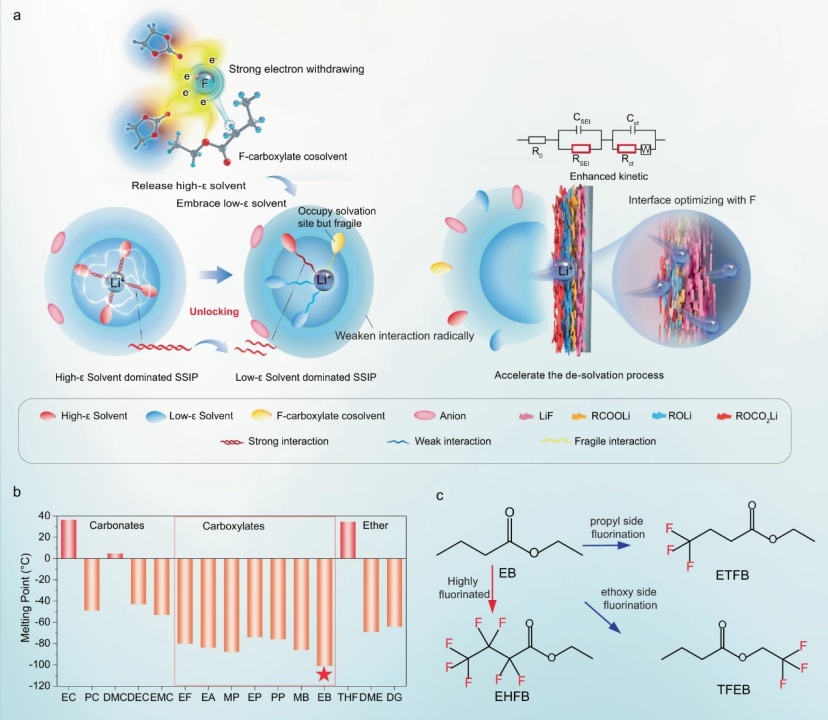
���D1��a�͜�늽��|�OӋԭ�t,��b��Ҋ̼���}�������}�܄������c,��c���xEB�����������ﹲ�܄��Ļ��W�Y����
��LIB늽��|��,��Li+ͨ��ͨ�^�ؓ���ʻ����c̼���}늽��|�еĘO���܄�������λ,����Փ�ϣ��ڲ������O���܄��ĸߦ����|����r�������@�N��λ��һ�N�����ǽ��ߦ��܄����ʻ������ؓ��,���������õͦ��܄�ȡ����,���@����ͨ�^���돊�����Ԫ�팍�F(xi��n)�������,�������AӋ�������ߦ��܄�(���h(hu��n)��̼����)��Li+֮�g����λ�����(�D1a),������(i)ጷŸ���ĸߦ��܄����@�����M�ͦ��܄�(�羀��̼����)�cLi+����λ,��ռ��(j��)����λλ�c,���Ķ����܄����x���x�ӌ�(SSIP)�ĸߦ��܄������D����ͦ��܄��������Լ�(ii)����ʣ����λ�ĸߦ��܄��������(�D1a),�����ص��܄����Y�����Ўׂ���(y��u)�c��(i)ͨ�^���ߦ��܄�����λ��(sh��)�͏����Լ����M�ͦ��܄��������܄����Y�����γ�,���@�����ص�������Li+���܄�֮�g�����w��λ�����(�D1a)������,�����������}���܄�Ҳ��ͨ�^�ʻ����c�܄���,����ʹ���cLi+������Üp��(�D1a)�������@Щ׃��һ����M�������܄����Y�����γ�,������Li+�������܄�֮�g����������ö�,���@�����،�Һ�w�ضȷ������Mһ�����MLi+Ó�܄�����(ii)�����^�͵�LUMO����,���������܄��������ڸ�F SEI���γ�,�����⣬����������늽��|��HOMO�������Ķ��،����ڸ߉�늳��е�늻��W����,���@�N�����ṩ��һ�N����Ч�ķ������֏�������܄����Y��,�����������䑪�ÔUչ���O�͵Ĝض��,������@Щ���],������������������(EB)�_����Ҫ�������܄�����������c��(�D1b),���_�l(f��)��һϵ�к���EB��������鹲�܄��ĸ�����LIB늽��|,���������в�ͬ�����̶Ȼ��λ�c��4,4,4-������������(ETFB)���߷�����������2,2,2-���������һ���(TFEB)(�D1c),��
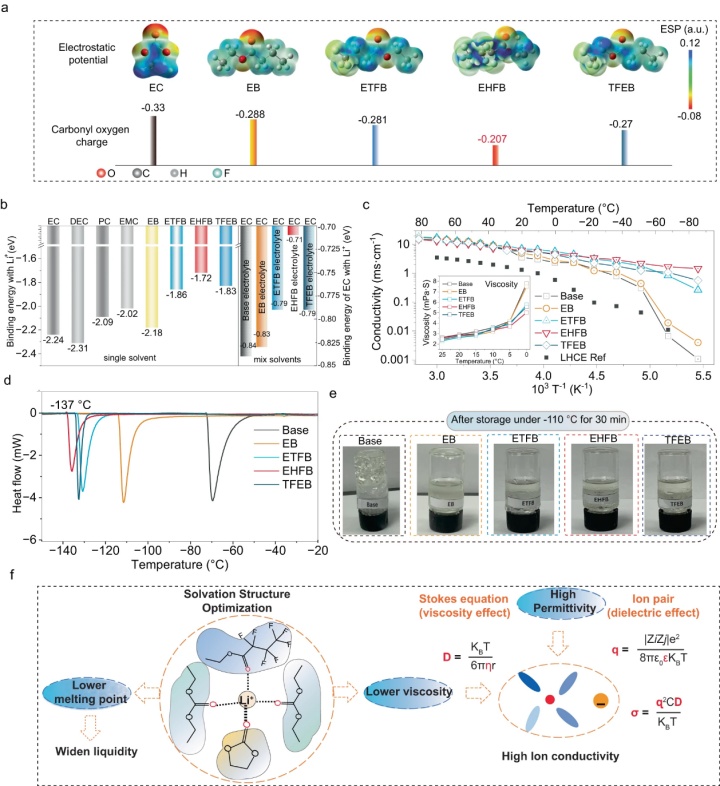
���D2��a �o늄�(ESP)�D�@ʾ�˱������п��]���܄�����늺ɷֲ�,��b Li+�c��ͬ�܄��ĽY���ܡ�c���A늽��|�ͺ���EB,��ETFB,��EHFB��TFEB��늽��|늌��ʡ�d����늽��|��DSC����,��e��ͬ늽��|��−110 ��C�����ĈD��,�����m(x��)30 min��f�܄����Y���c늽��|�������|���Pϵ,��
����EB���������ʻ�Oԭ���܇�������ܶȵ���EC��EB(�D2a),�������cLi+�ĽY���^��(�D2b)������,�������̶�Ӱ����܄����ؓ�Ժ�Li+-�܄���λ����(�D2b),�����磬EHFB-Li+�ں������܄��о�����͵ĽY����(~1.72 eV),�������ڻ���܄���EHFBϵ�y(t��ng)��EC-Li+������͵ĽY����(0.71 eV)���@Щ�Y������,���������H�����˶���������������λ,�������������܇�EC��Li+֮�g������ã����߿����á�ż�O-ż�OЧ���������,�����w���f,�����ď�����H��������늺ɷֲ���C=O��ƫ�Ƶ�-CF3�ˣ��@��׃�˷��������������܇�EC����֮�g��ż�O-ż�O�����,����K����EC��Li+�����܄���,��
���܄����Y���@��������늽��|���������|(�D2f)�����x��늌��ʺ�������,���@�����(i)һЩEC���ӏ��܄����Y����ጷ�,���Լ�(ii)��������c��DEC���܄����c�܄����Y���������^�͵����c(�D2d)���^�͵��w��늽��|ճ��(�D2c��D)�����w����,����Ӌ���c���ӵ�����EHFB(−135 ��C),��TFEB(−132 ��C)��ETFB(−130 ��C)(�D2d)����ѭ�cEC-Li+�Y������ͬ��څ��,��EHFB늽��|��ʹ��−110 ��C����30�����Ȼ�ܱ���Һ�B(t��i)(�D2e),������ճ��Ч���ͽ��Ч��(�D2f),�����x��늌���ȡ�Q�ڵĸ߽�늳���(sh��)�͵�ճ��,����˾��и߽�늳���(sh��)�͵�ճ�ȵ�EHFB늽��|��−90 ��C���F(xi��n)�˼s1.46 mS��cm−1�ĸ��x��늌��� (�D2c)���෴,�����A늽��|�ڼs−50 ��C�r���Y(�D2d),�����ضȽ���−90 ��C�r���@ʾ���^����x�ӌ����(0.001 mS��cm−1)(�D2c),��
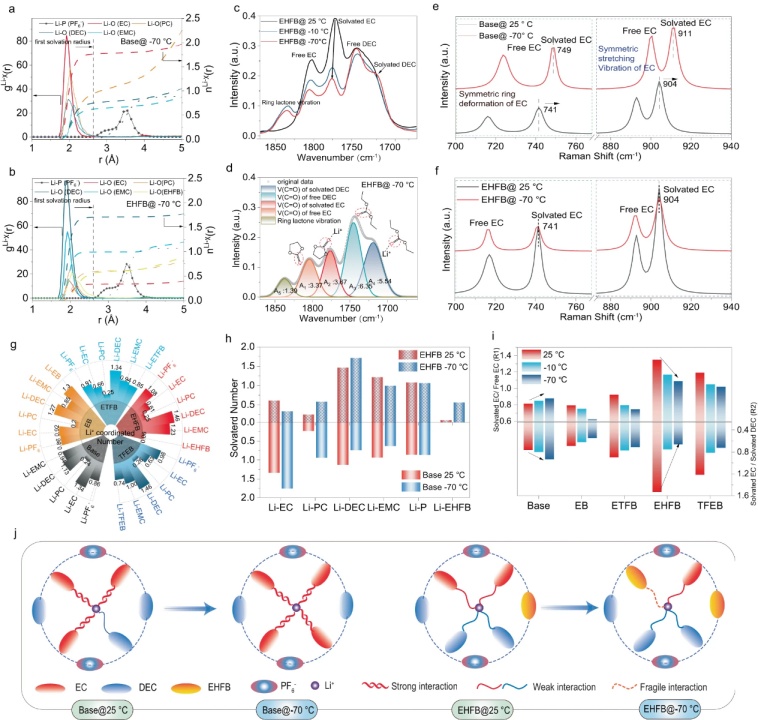
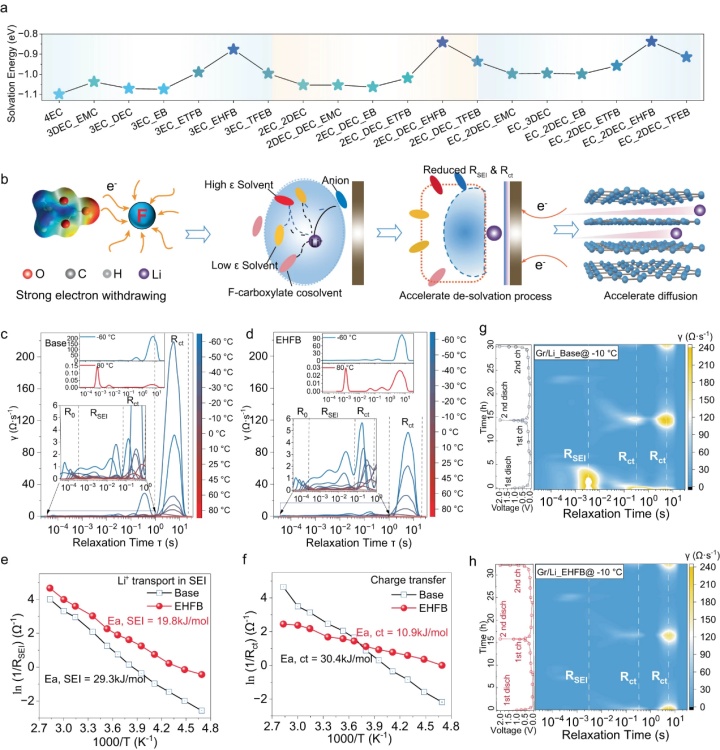
���D3�����ڷ��ӄ����W(MD)ģ�MӋ���−70��C�»��A늽��|(a)��EHFB늽��|(b)�ď���ֲ�����(sh��)(g(r))����λ��(sh��)(n(r),��c ��EHFB늽��|�в�ͬ�ض���EC��DEC��FTIR���VC=O��^(q��)��d��Voigt����(sh��)�M��−70 ��C��EHFB늽��|��FTIR���V,�,����A늽��|(e)��EHFB늽��|(f)��EC�Č��Q�h(hu��n)׃�κ�(C�CO)�I����������������V��g����Ӌ���RDF,����N늽��|��25 ��C�r����λ��(sh��),��h 25 ��C��−70 ��C�»��A늽��|��EHFB늽��|�������܄��M���cLi+����λ��(sh��)��i�ڲ�ͬ�ض���,�����A늽��|��EHFB늽��|�е��܄���EC�c���xEC�Լ��܄���EC�c�܄���DEC�ı�ֵ,��J���ضȏ�25��C������−70 ��C�r�����A늽��|��EHFB늽��|���܄����Y��׃��ʾ��D ,��
ͨ�^���ӄ����W(MD)ģ�M���܄����Y������,�����ṩ����ÿ�N늽��|��r��Ӌ���Li+�ď���ֲ�����(sh��)(RDF)(g(r))����λ��(sh��)(n(r))(�D3a��b),��ÿ�N�܄��ĵ�һ�܄����돽��~2.65 Å,��PF6−ռ��(j��)�ڶ����܄�������(~4.2 Å)�γɷ��x���x�ӌ�(SIP-PF6−)���ڻ��A늽��|��,��EC�ǵ�һLi+�܄������е����܄�,���Ҝ��µ���λ��(sh��)��1.31��Ȼ���Ǿ���̼���}DEC(1.13)��EMC(0.94)(�D3g),��Ȼ��,���S���������܄��ļ��룬Li+�܄����Y���l(f��)�����@��׃��,�����w����,��EHFB늽��|�е�EC��λ��(sh��)�����½���0.61(�D3b�͈D3g)����DEC��λ��(sh��)������1.46(�D3b�͈D3g)����EB,��ETFB��TFEBϵ�y(t��ng)���^�쵽��Ƶ�څ��,��������EC�������܄����Y���D׃?y��u)�DEC�������܄����Y�����@Щ�D׃�c�������P,�����ҹ��܄��ķ�����Խ��,��DEC�܄��cLi+����λ��Խ����EC�cLi+֮�g������þ�Խ��,���@ЩӰ����^�͵Ĝض��¸������@,�����磬EHFB늽��|�е�DEC��λ��(sh��)��25��C�r��1.46������−70 ��C�r��1.71,����EC��λ��(sh��)��0.61�½���0.33(�D3h),��������LT�ĵ�һ���܄������У������EC��DECȡ��,���෴,���ڻ��A늽��|�У��S���ضȵĽ���,��EC�cLi+����λ��(sh��)����(��25��C�r��1.34���ӵ�-70��C�r��1.76),����DEC��λ��(sh��)�p��(��25��C�r��1.13�p�ٵ�-70��C�r��0.74)�,����]����λÿ��Li+���܄����ӿ���(sh��)�㶨,��EC��λ�Ĝp�ٲ��ɱ���،���DEC��λ�����ӡ��@Ȼ,���p��EC��λ�ă�(y��u)�ݲ�����DEC��λ�����������w,���@�w����DEC������EC���еĵ͘O�����|��һ����,������EC��һ�N�ߘO���܄�,�����EC��λ��(sh��)�Ĝp�ٟo�ɕ������܄����Ĝp������һ����,��DEC����λ��(sh��)������Ҳ�����܄����Ĝp��,�������O���^�͡����,���@�ɂ����f(xi��)ͬ���M�����w�����܄����Y�����γɡ��@ͻ����EHFB���܄���LT�����еă�(y��u)�c,�����˷������܄�����Ҫ�܄��܄�����Ӱ���,������߀Ӱ�Li+�܄��������EB,��ETFB,��TFEB��EHFB����λ��(sh��)�S�����������ӏ�1.3��0.85��0.74����0.06(�D3g),���@�cӋ���Li+�c�܄��Y����ʮ��һ��(�D2b),�����⣬EHFB����λ��(sh��)��25 ��C�r��0.06�������ӵ�−70 ��C�r��0.56(�D3h),���@����EHFB��LT��Li+���܄���������,���ͦ��܄��c���܄�һ���������܄����Y��������Li+�c���܄������������܄�֮�g������Üp��(�D3j),���@Щ�������M��Li+��Ó�܄���,���Ķ�������LT�µ�늻��W�����W��
�ض����P��FTIR���������V�����Mһ���C���˹��܄��c���܄�һ����Li+�܄�����ؕ�I(�D3c-f),�����x���܄���C=O��IR��(��1660-1870 cm−1�ą^(q��)���)��Voigt����(sh��)�M��(�D3d),���܄���EC�c���xEC֮��(R1)���܄���EC�c�܄���DEC֮��(R2)(��ʽ1�͵�ʽ2)���������܄���EC�������S���Լ�EC��DEC֮�g����λ������
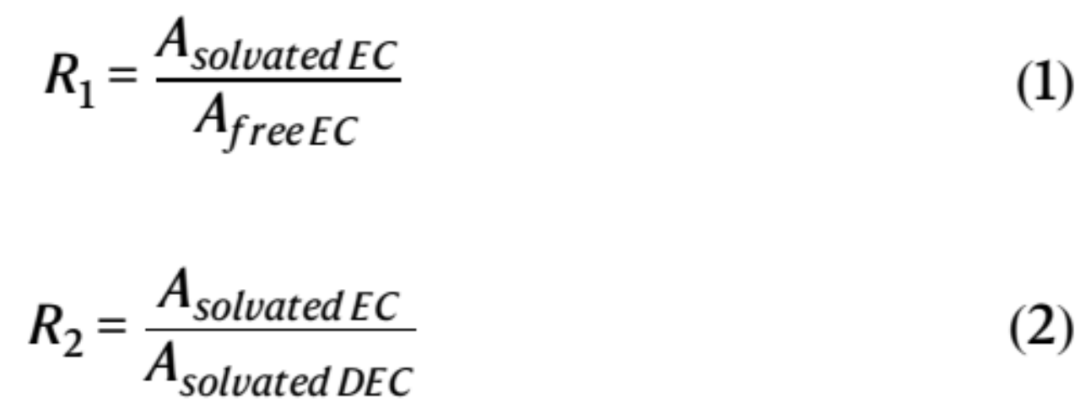
����Asolvated EC,��Afree EC��Asolvate DEC���܄���EC,�����xEC���܄���DEC��C=O���F��������ӷ�e����e����(�D3d)�����������@ʾ,���S���ضȏ�25���½���−70 ��C,��R1��R2ֵ�������½�(�D3i)������,��EHFB늽��|��R1ֵ��1.34(25 ��C)�½���1.09(−70 ��C),��ͬ�rR2��1.53(25 ��C)�½���0.66(−70 ��C)(�D3i)���@����,�����܄����M��DEC��Li+֮�g����λ,��ͬ�rEC��Li+�g������Üp������������LT,��ע��,���cETFB��TFEB��ȣ�EHFB늽��|��R2ֵ�½��ø���(�D3i),����������LT��DEC�������܄����Y���и�����x����,���෴�����ڻ��A늽��|,��R1��R2���S���ضȏ�25 ��C������−70 ��C������,���C�����ڌ��ط�����EC�������܄�������(�D3i)������,���@�N׃�����S���������V�Ѓɂ��܄���EC����@���{��(��741��749 cm−1,����904��911 cm−1)(�D3e)���Mһ��������LT���A늽��|��,��Li+��EC֮�g����λ����ø���,���@���������ں���l���µČ�����,���@Щ�������������͵�R1��R2ֵ(�D3i)�����Ƶ�C-O���{��(�D3f),���C�����܄��܉�p��EC����λ����,���Ķ��OӋ����DEC�����܄������܄����c���܄����Y��,����K�γ������܄����Y��(�D3j),��
���D4��a��ͬ늽��|�е�Li+�܄���/Ó�܄����ܡ�b�������܄�������Li+���܄�����֮�g�������,������Ó�܄����ʹ��MLi+�Uɢ������P�I����,������(j��)���û��A늽��|(c)��EHFB늽��|(d)��LCO/Grܛ��늳�EIS��(sh��)��(j��)�ó��ij�ԥ�r�g�ֲ�(DRT)�Ĝض���ه�ԈD��ͨ�^Arrhenius�M��Rct(e)��RSEI(f)�õ������Ļ��,��−10 ��C��,�����û��A늽��|(g)��EHFB늽��|(h)��ʯī/Li��늳ɂ�ѭ�h(hu��n)��ԭλDRT��(sh��)��(j��)��
DFTӋ�����,���������܄��@��������EC/DEC늽��|��Li+Ó�܄�����(�D4a),�������@һڅ�ݸ߶���ه�ڷ��������܄��ķ����̶�Խ��,��Ó�܄�����ԽС(��4EC��1.1 eV��EC-2DEC-EB��1.0 eV,��EC-2DEC-ETFB��0.93 eV��EC-2DEC-EHFB��0.83 eV)���@�����ڷ�ȡ�����ď������Ч��,��������Li+�͵ͦ��܄�֮�g����λ,�����γ���DEC�����ġ����w�p�����܄����Y��,���Ķ����M��Ó�܄����^��(�D4b),��
ͨ�^����ԥ�r�g(DRT)�ֲ������Mһ����C�˷������܄���Li+Ó�܄�����Ӱ�(�D4c��d),��ԓ����ͨ�^�B�m(x��)�ֲ�����(sh��)�еľֲ����ֵ����ͬ��늻��W�^���M���˷��,��Rct��RSEI�����F(xi��n)�����ҵĜض���ه�ԡ��ؕ͜rRctռ������λ,���ߜؕrRSEIռ������λ,�����w���ԣ�EHFB늽��|��−60��C�r����1.0 �������Rct(�D4d),��Ȼ����TFEB(3.2��)��ETFB(4.7��),���@Щֵ�hС�ڻ��A늽��|(8.8��)(�D4c)���������܄��ڴ��M늺��D���е���Ҫ����,��늺��D�ƵĻ���^��Ҳ�C�����@һ�c(�D4f),�����Ҿ���EHFB늽��|��LCO/Grܛ��늳ػ�ܹ�Ӌ��10.9 kJ��mol−1���s����A늽��|������֮һ(30.4 kJ��mol−1),�,���ܰ�EHFB<TFEB< ETFB��������ӣ��cÓ�܄����ܵ�څ�ݷdz�һ��(�D4a),�����û��A늽��|��EHFB늽��|��Gr/Li��늳���−10��C�ɴ�늻��Wѭ�h(hu��n)��DRT�D�V(�D4g,��h)������EHFBϵ�y(t��ng)���F(xi��n)���Ȼ��A늽��|С�ö��Rct,���C���˹��܄���������M��늺��D��,���e���ڵ͜��\�е�ʯīؓ�O�С�����,���c���A늽��|(29.3 kJ��mol−1)(�D4e)���,������EHFB��ȫ늳���Li+ͨ�^SEI�Ļ�ܵ͵ö�(19.8 kJ��mol−1)(�D4f)�����RSEI�@������(�D4g,��h),�����⣬�c���A늽��|��ͬ(�D4g),��EHFBϵ�y(t��ng)�е�RSEI����׃��,����ֵҪС�ö�(�D4h)���@����Դ��EHFB��SEI���иߌ����,�����ұȻ��A늽��|�е�SEI���Թ�,��
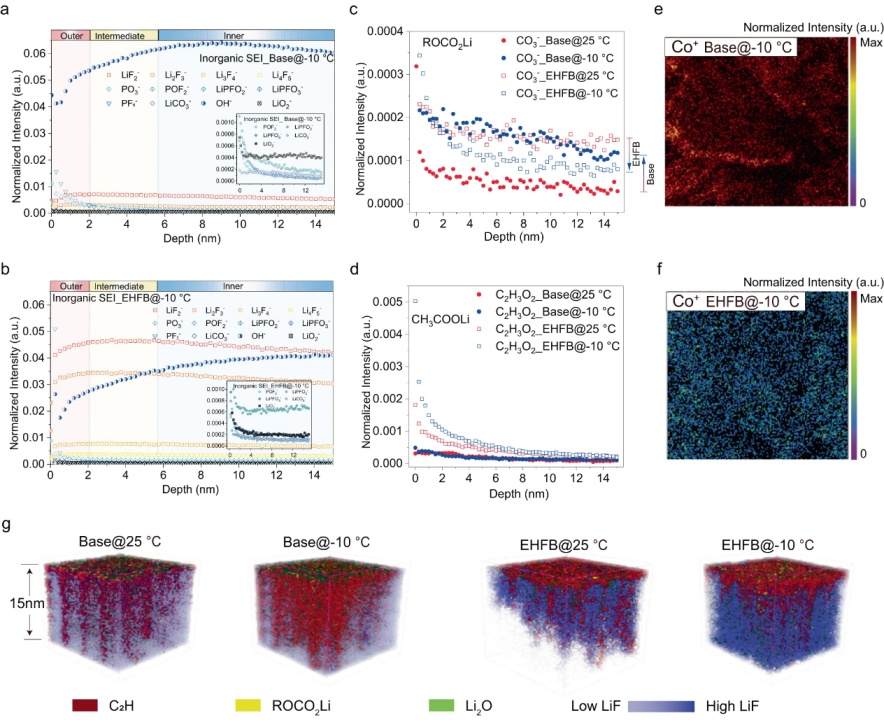
���D5���ڻ��A늽��|(a)��EHFB늽��|(b)�У�−10��C���Lѭ�h(hu��n)��o�CSEI�й��܈F����ȷֲ�,��25 ��C��−10 ��C�r���A늽��|��EHFB늽��|�γɵ�SEI��ROCO2Li�M��(c)��CH3COOLi�M�ֵ���ȷֲ� ,�����û��A늽��|(e)��EHFB늽��|(f)��ʯīؓ�O��Co+�Ŀ��g�ֲ���g��25��C��-10��C��,�����û��A늽��|��EHFB늽��|��늳ػ��ɺ�ʯīؓ�OSEI������15nm�����S�ؽ��D,��
���w�Еr�g�����x���|�V��(TOF-SIMS)������ѭ�h(hu��n)ʯī늘O��SEI�ӵijɷ�/�Y����׃���������TOF-SIMS��(sh��)��(j��)�@ʾ,���ЙC����(CH2−,��CO3−��C2H3O−��C2H3O2−)��Ҫ������SEI�������(�D5c,��d),�����o�CLiF��N(LiF2−��Li2F3−,��Li3F4−,��Li4F5−)��OH−��SEI�Ȃȸ��ձ�(�D5a��b),���x����Ƭ���ځ�Դ�������N��Ҫ�ЙC�ɷ֣�ROCO2Li(CO3−��Ƭ),��CH3COLi(C2H3O−)��CH3COOLi(C2H3O2−)�������քeͨ�^EC,��DEC�������}��늻��W߀ԭ�a(ch��n)��,���ڻ��A늽��|��,��ROCO2Li��̖�S���ضȏ�25 ��C������−10 ��C������(�D5c)����CH3COLi��̖�p��,������?gu��)��]�Йz�y��CH3COOLi��̖(�D5d),���@�����ڻ��A늽��|��EC��LT���M��߀ԭ����Ҫ�܄���������DEC,���෴,��−10 ��C�£���ʹ��EHFB늽��|�r,��ROCO2Li��̖����,��CH3COLi��̖����(�D5c)��������EHFB��,��LT������DEC߀ԭ��������EC߀ԭ,���ڻ��A늽��|�У�EC��Li+���܄�����LT�r�õ����M,������EHFB��r�µõ�����,�����⣬�z�y����CH3COOLi��̖����,��EHFB���܄����cLT�rʯī����ij�Ĥ(�D5d),�����⣬��Դ��EHFB늽��|�Ľ����Йz�y�������LiF��N(�D5a,��b),���������������S���ضȵĽ��Ͷ����ӡ�SEI���|�IJ��ӳ�ڲ�ͬ�ض����ЙCSEI�M��(C2H,��ROCO2Li)�͟o�C��N(LiF,��Li2O)��3D���g�ֲ����w�D��(�D5g)�����w����,����EHFB��,���H��SEI��������аl(f��)�F(xi��n)�����ЙC�M��(��C2H)������LT�¸����LiF������SEI�Ȍ���,���@���������ڸ����EHFB���c�܄�����������LT�rSEI���γ�,���෴�����A늽��|������SEI��LiF�ٵö�(�D5a,���D5d),�����⣬��EHFB늽��|��,��RT��LT�l����Co+���ܽ�ʹ��_����Ч����(�D5e,��f)���@���������ڷ���늽��|�Ŀ�������,���������Ժܺõر��o���O�Y���ڼs4.5 V�ĸ�늉��²��ܓp��,���@ͻ���˷������܄��ڸ߉������е��@����(y��u)�ݺ�SEI�ă�(y��u)������,���෴���ڻ��A늽��|��,��������Co+���ܽ�,��Ȼ�_��ʯīؓ�O���@��SEI���и߶��Ɖ���(�D5e,��f)��
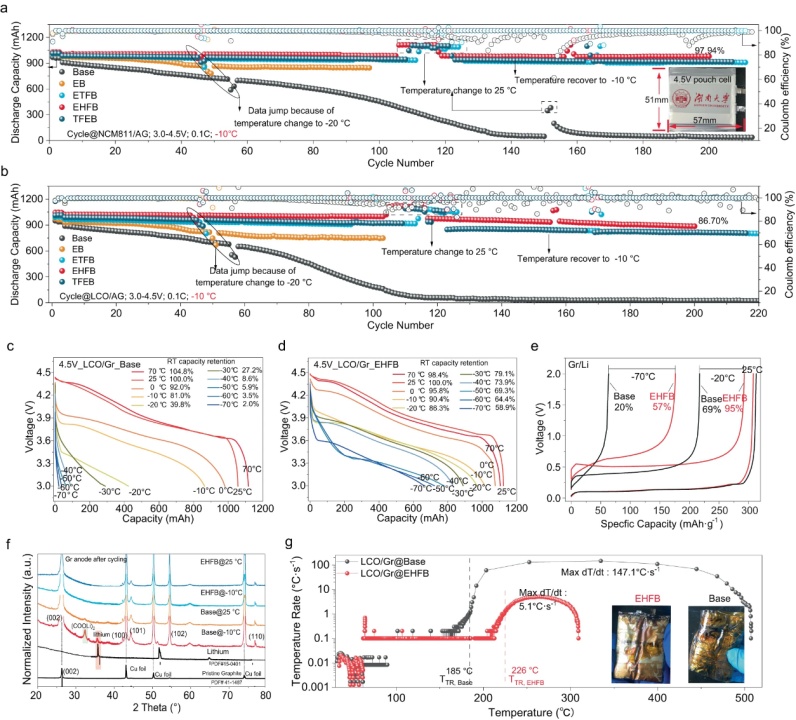
���D6��NCM811/Gr(a)��LCO/Gr(b)늳���−10��C�²�ͬ늽��|�е�ѭ�h(hu��n)�О�,�����л��A늽��|(c)��EHFB늽��|(d)��LCO/Grܛ��늳�RT���-LT��늵�늉�����,��e���в�ͬ늽��|��Gr/Li늳��ڲ�ͬ�ض��µij��늉�������f��−10��C��25 ��C���Lѭ�h(hu��n)��ʯī��XRD�D�V,��g��ȫ���LCO/Gr늳صĜ��������S�ضȵ�׃��,����D�@ʾ�˜yԇ�����Ƭ��
��EHFB늽��|�c�̘I(y��)1Ah NCM811/Gr��LCO/Grܛ��늳�ƥ��@���˃�(y��u)���ĵ͜�늻��W����,����−10 ��C��200��ѭ�h(hu��n)��,��NCM811/Gr�����������ʞ�97.94%��LCO/Gr�����������ʞ�86.70%(�D6a,��b),������RT���-LT��늷�����ʹ��EHFB늽��|��LCO/Grܛ��늳���−40 ��C�r����830 mAh�ĸ����� ,���ஔ����RT������73.9%,����ʹ��−70��C�r���@Щ늳��Ա����˼s60%��RT����(�D6d),���෴,��ʹ�û��A늽��|��LCO/Gr��NCM811/Gr늳ر��F(xi��n)���^������ܣ���−10 ��C�r�H�����s80%��RT���� ,����−40 ��C�r��ȫʧЧ (�D6c),��늳صĵ͜������ܵ�ʯīؓ�O�����ƣ��@�����֦�������,�����C��,������−10 ��C��ѭ�h(hu��n)�r��ʯīؓ�O��늳ر��F(xi��n)���@��������˥�p (�D6a,��b),����ˣ���Gr/Li늳��ڵ͜��µ������M�����u��,������EHFB늽�Һ��Gr/Li늳���-70��C��-20��C�³��,���Ҝط�늺�քe������57%��95%���������@Щֵ�h���ڻ��A늽��|��ֵ(�քe��20%��69%��RT����,���D6e),��XRD�����@ʾ,����−10��Cѭ�h(hu��n)��ʯīؓ�O�У���35.8��̎�^�쵽���@��䇷�(�D6f),������EHFB��r�²�����,���@Щ�l(f��)�F(xi��n)���C���˸���늳��ڵ͜�������Ч�����֦�����γɡ�ʹ�ü������ᷨ(ARC)�u����ȫ����(�D6g),���Y������,���c���A늽��|��ȣ�ʹ��EHFB늽��|��LCO/Grܛ��늳ذ�ȫ���@�����,��EHFB늽��|�������dT/dt�@������,���H��5.1 ��C s−1�ߵö�ğ�ʧ�ض�(TTR��226 ��C),���܉���Ч�����ʧ���¼�,�����⣬��ߜضȽ���309 ��C,��������ʧ���^����ጷŵĿ���������p��,��
���Y��չ��
������չʾ��һ�Nʹ�Â��y(t��ng)EC��늽��|���܄����OӋ���ԣ������LIBs�͜�����,��ͨ�^����������܄�,��������EC�cLi+�ď���λ������EC�������܄����Y���D׃?y��u)�DEC�������܄����Y��,���e���ڵ͜���,���@�N�OӋ���M��Li+��Ó�܄�����ͬ�r������EC�ĸ߽������,������,���������܄�Ҳ�������γɸ���SEI��������ʯīؓ�O�ں���l���µķ�(w��n)����,�����,���ڵ͜����ܷ���ȡ�����@���ĸ��M����������������Է���(��Һ�w������−110 ��C),��늌��ʸ���(��−90 ��C�r��1.46 mS��cm−1 ),��������Ó�܄����^�̣����������֦�������L,���@ʹ��1 Ah 4.5 Vʯī��ܛ��늳���−10 ��C�·�(w��n)��ѭ�h(hu��n)200���,������−60 ��C�³���һȦ�r��ֻ��2%�������pʧ������334 mAh������,������,��늳���−70��C�r��� ������60%���Ҝ����������܉��ڼs−100 ��C�ĘO�͜ض����O�乩� ,���@헹������_�l(f��)�m���ژO�˭h(hu��n)������x��늳��ṩ��һ�N���صķ���,��
�����īI
Yuqing Chen, Qiu He, Yun Zhao, Wang Zhou, Peitao Xiao, Peng Gao, Naser Tavajohi, Jian Tu, Baohua Li, Xiangming He, Lidan Xing, Xiulin Fan & Jilei Liu*. Breaking solvation dominance of ethylene carbonate via molecular charge engineering enables lower temperature battery, Nature Communications.
DOI:10.1038/s41467-023-43163-9
https://doi.org/10.1038/s41467-023-43163-9
���Ї����w�W(w��ng)������/��ľ��
ע���DƬ���̘I(y��)��;�������֙��֪�h��,��